AI Insight
The STRENGTH trial (NCT05386680) evaluated the safety and tolerability of intrathecal onasemnogene abeparvovec (OAV101 IT) in 27 patients with spinal muscular atrophy (SMA) aged 2 to under 18 years who had previously received nusinersen or risdiplam. All participants experienced at least one adverse event, with the most common being nasopharyngitis, pyrexia, and vomiting; serious adverse events occurred in 14.8% of participants, primarily respiratory infection-related, and no deaths or study discontinuations due to adverse events were reported. The safety profile of OAV101 IT in this treatment-experienced population was consistent with that observed in treatment-naive patients, suggesting no markedly increased risk associated with prior SMA therapy exposure.
Why it matters
Many SMA patients initiated treatment with approved therapies before gene therapy became available, and this study provides critical safety data to support the potential use of OAV101 IT as a switching option for patients who have not achieved sufficient benefit from existing treatments. Demonstrating a manageable safety profile in this broader population could meaningfully expand access to one-time gene replacement therapy for a larger proportion of SMA patients.
Understand the Science
Abstract
Intrathecal onasemnogene abeparvovec (OAV101 IT) may enable a one-time gene transfer therapy, addressing an unmet need across the broader spinal muscular atrophy (SMA) population. STRENGTH (NCT05386680) was a 52-week, phase 3b, single-arm, open-label, multicenter study evaluating OAV101 IT in participants with SMA aged 2 to <18 years who discontinued nusinersen or risdiplam. The primary objective was safety/tolerability. Twenty-seven participants were enrolled (mean (s.d.) age at OAV101 IT, 7.4 (3.35) years; range, 2.4–17.7 years) with prior nusinersen or risdiplam (mean (s.d.) duration of 4.3 (1.07) years; range, 1.86–6.18 years or 3.0 (2.02); range, 0.39–6.28 years). All (n = 27) experienced at least one adverse event (AE), most frequently nasopharyngitis (n = 15, 55.6%), pyrexia (n = 14, 51.9%) and vomiting (n = 13, 48.1%). Thirteen participants (48.1%) experienced treatment-related AEs, most frequently vomiting (n = 6, 22.2%), headache (n = 4, 14.8%) and pyrexia (n = 3, 11.1%). Serious adverse events (SAEs) were reported for four participants (n = 4, 14.8%) (mostly respiratory infection related). No AEs leading to death or study discontinuation were reported. Adverse events of special interest (AESI) reported were in categories of transient thrombocytopenia (n = 8, 29.6%), hepatotoxicity (n = 4, 14.8%) and signs and symptoms that may be suggestive of dorsal root ganglia toxicity (n = 2, 7.4%). The OAV101 IT safety profile was consistent with findings in treatment-naïve patients. Trial registration: ClinicalTrials.gov identifier: NCT05386680.
Similar content being viewed by others


Main
SMA is a rare, autosomal recessive neuromuscular disease caused by biallelic loss or mutation in the SMN1 (survival motor neuron 1) gene on chromosome 5q13, which leads to intracellular survival motor neuron (SMN) protein deficiency, motor neuron dysfunction and degeneration and progressive muscle weakness1,2,3. The SMN2 (survival motor neuron 2) gene, a partially functional paralog of SMN1, produces low levels of SMN protein. The severity of the disease is mainly influenced by the number of copies of the SMN2 gene1. Three disease-modifying treatments are currently approved for patients with SMA. Nusinersen, an intrathecally administered antisense oligonucleotide, and risdiplam, an orally administered small molecule, are SMN2 splicing modifiers that increase the amount of available functional SMN protein4,5. Onasemnogene abeparvovec is a one-time, weight-based, intravenous, adeno-associated virus 9 (AAV9) vector-based gene transfer therapy that delivers a stable, fully functional human SMN transgene into cells6,7. Onasemnogene abeparvovec is approved for patients with SMA aged younger than 2 years in the United States and in Europe with recommended doses for patients weighing up to 21 kg6,7. Other countries have differing approvals that vary by age and/or SMN2 copy number.
Despite the availability of these treatments, there is a need for treatment options for the broad patient population with SMA. For example, some patients may be too old or too heavy to be eligible for treatment with intravenous onasemnogene abeparvovec, or some patients may want to discontinue their current treatment because of either lack of continued benefit or tolerability or challenges with access to ongoing dosing8,9.
Although patients with SMA may be diagnosed by newborn screening, if available, or have symptoms that appear in infancy, those with a later onset of disease may not be identified until they are beyond the window of approved intravenous onasemnogene abeparvovec treatment10. OAV101 IT may be a potential one-time treatment option for a broad range of patients with SMA for whom repeated doses may be challenging9,10. OAV101 IT would enable a fixed-dose administration of onasemnogene abeparvovec, allowing for reduced viral load and reduced systemic vector exposure compared to weight-based, systemic intravenous dosing, but with similarly sustained SMN protein expression and durable effects as a result of the mechanism of action at the target site11,12.
The phase 3, randomized, double-blind, sham-controlled STEER (NCT05089656) study evaluated OAV101 IT in a treatment-naïve patient population with SMA (aged 2 to <18 years) over 52 weeks. STEER demonstrated an increase from baseline in motor function based on the Hammersmith Functional Motor Scale–Expanded (HFMSE) for participants who received OAV101 IT (n = 75) compared with sham control (n = 51) with a favorable benefit−risk profile and similar incidence rates of adverse events (AEs), serious adverse events (SAEs) and adverse events of special interest (AESI) reported for both OAV101 IT and sham groups13.
Although STEER provides safety and efficacy evidence for OAV101 IT for treatment-naïve participants with SMA, it is likely that most patients who receive OAV101 IT in clinical practice will be treatment-experienced patients who are receiving (or have previously received) nusinersen or risdiplam. The STRENGTH study (NCT05386680) is, to our knowledge, the first study to evaluate the safety, tolerability and efficacy of OAV101 IT for participants with SMA who discontinued previous nusinersen or risdiplam treatment.
Results
Participants
Participants were enrolled at 13 sites in nine countries. A total of 41 participants were screened (Fig. 1), of whom 14 did not meet eligibility criteria (for example, abnormal laboratory tests, infection or elevated anti-AAV9 titers). A total of 27 participants were eligible to receive OAV101 IT. The first participant’s first visit (screening) was on 12 January 2023 (treatment received on 22 February 2023), and the last participant’s last visit was on 29 November 2024 (treatment received on 29 November 2023). Two participants (7.4%) discontinued the study after OAV101 IT administration (day 106 and day 273) due to guardian decision (for example, travel logistics difficulties and worsening of symptoms). Results are reported for participants from the full analysis set (FAS) for assessment at each study visit. Participant data are missing if they did not complete an assessment. Four participants initiated add-on therapy after OAV101 IT administration, and these participants were included in the FAS.
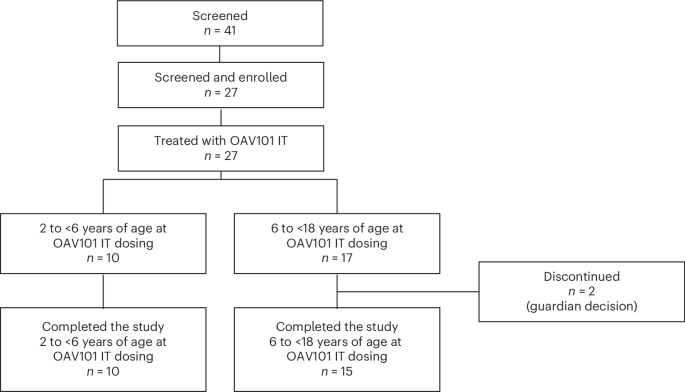
CONSORT diagram and patient disposition.
Baseline demographics and characteristics for the overall study population
All participants were symptomatic at the time of enrollment, and the most frequent symptoms were limb weakness (n = 26, 96.3%) and hypotonia (n = 19, 70.4%). Most participants (n = 23, 85.2%) had three copies of the SMN2 gene; 14.8% (n = 4) had two copies of the SMN2 gene.
The mean (s.d.) age at OAV101 IT dosing was 7.4 (3.35; range, 2.4–17.7) years (Table 1). All participants discontinued previous SMN-targeted treatments, including 21 participants who discontinued nusinersen (77.8%), four who discontinued risdiplam (14.8%) and two who had previously received both nusinersen and risdiplam (7.4%), although these treatments were not administered concurrently. The mean (s.d.) duration of treatment was 4.3 (1.07; range, 1.86–6.18) years for nusinersen and 3.0 (2.02; range, 0.39–6.28) years for risdiplam. Mean (s.d.) baseline HFMSE score for 26 participants was 24.3 (14.04; range, 2.0–46.5).
Primary endpoint
Safety
All participants (n = 27) experienced at least one AE, most frequently nasopharyngitis (n = 15, 55.6%), pyrexia (n = 14, 51.9%) and vomiting (n = 13, 48.1%) (Table 2). A total of 13 participants (48.1%) experienced AEs considered to be related to study treatment, most frequently vomiting (n = 6, 22.2%), headache (n = 4, 14.8%) and pyrexia (n = 3, 11.1%). SAEs were reported for four participants (n = 4, 14.8%), most frequently in the infections and infestations system organ class (n = 3, 11.1%) and most of which were respiratory infections (Table 2 and Supplementary Table 1). No AEs leading to death or study discontinuation were reported.
AESI reported were in the categories of transient thrombocytopenia (n = 8, 29.6%), hepatotoxicity (n = 4, 14.8%) and signs and symptoms that may be suggestive of dorsal root ganglia (DRG) toxicity (n = 2, 7.4%) (Table 2). The most frequently reported AEs in the transient thrombocytopenia category were epistaxis (n = 3, 11.1%) and contusion (n = 2, 7.4%) (Supplementary Table 2). These events were non-serious, mild or moderate in intensity and resolved. Two participants experienced gastrointestinal bleeding (one on the day of gastronomy surgery). These events were non-serious, mild or moderate in intensity and resolved within one day. No AEs were reported with the terms ‘platelet count decreased’ or ‘thrombocytopenia’.
Hepatotoxicity AESI included increased levels of liver enzymes (n = 4, 14.8%) (Supplementary Table 3) reported between 50 days and 87 days after OAV101 IT treatment for participants 2−10 years of age; events were mild to moderate in intensity, non-serious and resolved with prednisolone treatment. The mean (s.d.) duration of prednisolone treatment was 71.2 (34.40; range, 53–229) days (Supplementary Table 4). Based on the laboratory assessments, four participants (14.8%) had an alanine aminotransferase (ALT) increase more than 2× the upper limit of normal (ULN), including one participant (3.7%) who had an increase in ALT more than 10× ULN (heightened at week 42 but normal with retest) (Supplementary Table 5). No participants had ALT elevations more than 20× ULN. Two participants (7.4%) had aspartate aminotransferase (AST) more than 2× ULN, one (3.7%) of whom had AST more than 3× ULN. No participant had AST elevations more than 5× ULN. Total bilirubin did not exceed 2× ULN for any participants. No participants met Hy’s law criteria.
Signs and symptoms that may be suggestive of DRG toxicity included paresthesia in feet (n = 1, 3.7%) and sensory disturbance on forearms, neck and scalp (n = 1, 3.7%) (Supplementary Table 6). Paresthesia in feet occurred in a 4-year-old participant 28 days after OAV101 IT administration. This was considered to be related to OAV101 IT and was transient, mild and non-serious. The event resolved after 63 days without any treatment. Sensory disturbance on forearms, neck and scalp occurred in a 17.6-year-old participant 38 days after OAV101 IT administration, was considered not related to OAV101 IT, was transient, mild and non-serious and resolved within 3 days without any treatment. For both participants, neurologic examination was normal, and no additional tests were performed at the investigator’s discretion. No thrombotic microangiopathy, new malignancies or cardiac AEs were reported.
Secondary endpoints
Motor function
The mean (s.d.) change from baseline to week 52 in HFMSE score was 0.17 (2.89; range, −5.5 to 7.5), and the least squares mean (LSM) change from baseline to week 52 was 1.05 (s.e., 0.61; 95% confidence interval: −0.21 to 2.32; Cohen’s d: 0.36) (Table 3 and Fig. 2). The mean (s.d.) change from baseline to week 52 in revised upper limb module (RULM) score was 0.29 (2.85; range, −6.0 to 6.0), and the LSM change from baseline to week 52 was 0.59 (s.e., 0.55; 95% confidence interval: −0.56 to 1.73).
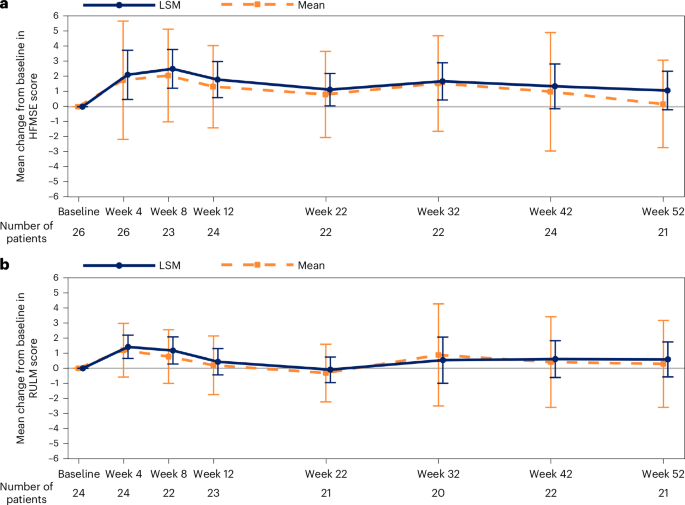
Data are displayed as mean (s.d.) and LSM (95% confidence interval). MMRM analysis accounted for missing data, and the remaining number of participants with data are reported for each timepoint. The mean (s.d.) change from baseline at week 52 was 0.17 (2.88) for HFMSE; LSM was 1.05 (95% confidence interval: −0.21 to 2.32, Cohen’s d: 0.36) after adjusting by baseline age stratum (LSM change from baseline to week 52, MMRM). The mean (s.d.) change from baseline at week 52 was 0.29 (2.85) for RULM; LSM was 0.59 (95% confidence interval: −0.56 to 1.73) after adjusting by baseline age stratum (LSM change from baseline to week 52, MMRM). MMRM, mixed model with repeated measurement.
Caregiver experience
Caregiver experience scores (assessment of caregiver experience with neuromuscular disease (ACEND)) remained stable over 52 weeks (Table 3). Mean (s.d.) change from baseline in ACEND instrument total score was 1.43 (9.32; range, −14.6 to 20.7); adjusted LSM change for ACEND was 1.06 (s.e., 1.92; 95% confidence interval: −2.90 to 5.02).
Exploratory endpoint
Motor milestones
Although some participants demonstrated new milestones at the week 52 visit, most demonstrated the same milestones as at baseline. At the week 52 visit, 17 of 24 participants (70.8%) who completed the study demonstrated milestones observed at baseline, and three of 24 participants (12.5%) demonstrated a new milestone (walking alone (n = 2) and standing with assistance and walking with assistance (n = 1)). Four participants did not demonstrate a milestone at the week 52 visit that had been observed at baseline (that is, baseline milestones: sitting without support (n = 2), hands-and-knees crawling (n = 1) and standing with assistance and walking with assistance (n = 1)). The participant who demonstrated hands-and-knees crawling at baseline but not at week 52 had sequelae from a broken arm. For the other three participants who demonstrated milestones at baseline but not at week 52, these milestones were demonstrated at other study visits.
Post-hoc analysis
HFMSE scores by age at symptom onset and visit prior to receiving add-on therapy
Post-hoc analyses of HFMSE change from baseline were performed for participants with age at symptom onset of 6 months or younger (n = 6) compared with participants with age at symptom onset after 6 months (n = 15). The change in HFMSE through week 52 demonstrated a consistent treatment effect irrespective of the age of symptom onset, with an HFMSE change from baseline of 0.33 in the group with symptom onset at 6 months of age or younger and 0.10 in the group with symptom onset at older than 6 months of age.
Four participants initiated add-on therapy after OAV101 IT administration and continued to be followed-up in the study. Post-hoc analysis indicated the LSM change from baseline to week 52 in HFMSE score when only including the visit prior to receiving add-on therapy was similar (1.02) to the LSM change from baseline to week 52 in the FAS (1.05).
Discussion
Overall, the safety profile of OAV101 IT for treatment-experienced participants was consistent with results from the STRONG (NCT03381729) and STEER (NCT05089656) studies in treatment-naïve participants. STRENGTH participants were previously treated with nusinersen (mean (s.d.), 4.3 (1.0); range, 1.9–6.2 years) or risdiplam (mean (s.d.), 3.0 (2.0); range, 0.4–6.3 years). No new safety concerns were identified in STRENGTH based on evaluation of AEs, SAEs or AESI. Of note, two participants had signs and symptoms that may be suggestive of DRG toxicity, in which both were non-serious, mild, transient and recovered without treatment.
At baseline, the population was heterogenous in terms of motor function based on HFMSE and RULM scores: the mean (s.d.) HFMSE score was 24.31 (14.0; range, 2.0–46.5), and the mean (s.d.) RULM score was 24.60 (7.4; range, 7.5–36.0). STRENGTH demonstrated stabilization in motor function and caregiver experience in treatment-experienced participants. The mean (s.d.) change from baseline to week 52 in HFMSE score was 0.17 (2.89; range, –5.5 to 7.5), and the mean (s.d.) change from baseline to week 52 in RULM score was 0.29 (2.85; range, −6.0 to 6.0). There is variability in change for individual participants, but overall stabilization was demonstrated as a group.
Because disease-modifying treatments for patients with SMA have been available in the United States and Europe since 2016, most patients who may seek treatment with OAV101 IT are likely to be treatment experienced. As such, STRENGTH was conducted in parallel with the STEER sham-controlled study for treatment-naïve participants. The STEER study demonstrated a statistically significant change from baseline in HFMSE score for OAV101 IT versus sham control over 52 weeks13. Although the results from STEER established the efficacy of OAV101 IT for treatment-naïve participants with SMA with no confounding factors of previous SMN-dependent treatments, efficacy data from STRENGTH for treatment-experienced participants with SMA are also important for clinicians. The importance of stabilization of motor function and caregiver experience is supported by one survey study (N = 822) in which most participants (97%) responded to indicate that stabilization was crucial and considered as therapeutic progress, with 81% considering it as major progress14.
Limitations of the present study include the open-label design and the lack of a matched control group, which may introduce potential for bias. In addition, the study did not have Cobb angle exclusion criteria, so progression in scoliosis and/or contractures may have impacted the demonstration of motor performance. At the time of study recruitment, many younger patients from participating countries had already received intravenous onasemnogene abeparvovec and were, therefore, not eligible for treatment with OAV101 IT. This resulted in most participants (17 of 27) being 6 years of age or older. During the study, four participants included in the FAS received add-on SMA therapy (nusinersen (n = 2), risdiplam (n = 1) and risdiplam followed by nusinersen (n = 1)) after OAV101 IT (initiated at week 21, week 33, week 44 and week 24 respectively). The LSM change from baseline to week 52 in HFMSE when only including participants with visits prior to receiving add-on therapy was similar (1.02) to the LSM for the FAS (1.05). Overall, add-on therapy initiated at the clinician’s discretion was safe and did not affect the conclusions of this study.
Conclusions
The STRENGTH study participants’ wide range of prior nusinersen or risdiplam treatment duration and baseline motor function was consistent with the broad range of characteristics observed in treatment-experienced patients with SMA. STRENGTH demonstrated a safety profile for OAV101 IT that was favorable and consistent with findings in treatment-naïve patients13,15. Therefore, OAV101 IT may be another treatment option for patients with SMA.
Methods
STRENGTH was undertaken in accordance with International Council for Harmonisation E6 Guidelines for Good Clinical Practice, with ethical principles in accordance with the Declaration of Helsinki. The study was approved by the institutional review boards (IRBs) at all participating institutions (University of Wisconsin Human Research Protection Program, Madison, WI, USA; McGill University Heath Centre Research Ethics Board, Montreal, Quebec, Canada; Commissie Voor Medische Ethiek UZ Gent, Ghent, Belgium; Comité de protection des personnes Sud Ouest et Outre Mer II, Toulouse, France; Comitato Etico Territoriale Lazio 3, Rome, Italy; Central Committee on Research Involving Human Subjects, The Hague, The Netherlands; Comité de Ética de la Investigación con medicamentos, Madrid, Spain; Tokyo Women’s Medical University IRB, Tokyo, Japan; Kurume University Hospital IRB, Kurume, Japan; The Royal Children’s Hospital Human Research Ethics Committee, Parkville, Victoria, Australia; Boston Children’s Hospital IRB, Boston, MA, USA; and Advarra, Columbia, MD, USA). Protocol approval was provided by IRBs, independent ethics committees and health authorities. Written informed consent was obtained from parents or legal guardians of enrolled participants. The STRENGTH study is registered at ClinicalTrials.gov (NCT05386680) and EudraCT (2021-006709-31).
Study design
STRENGTH was a 52-week, phase 3b, open-label, single-arm, multicenter study on the safety, tolerability and efficacy of OAV101 IT (1.2 × 1014 vector genomes) for participants 2 to <18 years of age who discontinued treatment with nusinersen or risdiplam. The study consisted of a screening period, which included two screening visits up to 45 days prior to treatment with OAV101 IT. Eligibility for enrollment was confirmed at baseline on day −1. On day 1, participants received a single IT administration of OAV101. Participants then entered the 52-week, open-label, follow-up period for safety and efficacy assessments (Supplementary Fig. 1).
Participants
Full eligibility criteria are described in the supplementary methods. Key inclusion criteria included genetic confirmation of SMA based on biallelic loss of function due to SMN1 deletion or mutations and any number of copies of the SMN2 gene, 2 to <18 years of age and having symptoms of SMA as defined in the study protocol. Participants needed to be able to sit without support and never walked independently. Participants must be diagnosed through newborn screening, or patients clinically diagnosed must have age of clinical symptom onset of younger than 18 months. Study participants must have received at least four loading doses of nusinersen or at least 3 months of treatment with risdiplam at screening; however, nusinersen must have been discontinued for at least 4 months, and risdiplam must have been discontinued for at least 15 days prior to baseline.
Participants were excluded if they had elevated anti-AAV9 antibody titers (>1:50), clinically significant abnormalities in test results during screening or contraindications for the lumbar puncture procedure. Participants were also excluded if they received vaccinations within 2 weeks prior to OAV101 IT administration, if they were hospitalized for a pulmonary event or for nutritional support within 2 months prior to screening, had inpatient major surgery planned, had an infection or febrile illness up to 30 days prior to OAV101 IT administration or required invasive ventilation.
Treatment
Approximately 24 hours prior to dosing, participants received oral prophylactic prednisolone (1 mg kg−1 d−1 up to a maximum of 60 mg d−1). Prednisolone was then administered for at least 30 days, including on the day of OAV101 IT administration. Tapering was then carried out at a rate of 0.20 mg kg−1 w−1 over at least 28 days, with dosages tapered down to ≤5 mg d−1 prior to stopping. OAV101 IT was delivered as a single injection. After OAV101 IT administration on day 1, participants remained in the hospital for at least 48 hours for monitoring.
Safety objectives
The primary objective was safety, assessed by the incidence of AEs, related AEs and SAEs, along with the number of participants with AESI, including hepatotoxicity, transient thrombocytopenia, thrombotic microangiopathy, cardiac AEs, signs and symptoms that may be suggestive of DRG toxicity and new malignancies. Laboratory assessments included those for liver function, including total bilirubin, AST, ALT and alkaline phosphatase (ALP). Potential Hy’s law cases (defined as ALT or AST more than 3× ULN and total bilirubin more than 2× ULN (mainly conjugated fraction) without notable increase in ALP to more than 2× ULN) were evaluated.
Efficacy objectives
Secondary objectives included efficacy assessments of motor function as measured by the change from baseline to week 52 in HFMSE and RULM scores.
The HFMSE is an SMA-specific, 33-item functional scale of motor skills administered by a qualified clinical evaluator16,17,18. Each motor skill item is scored on a three-point Likert scale from 0 (no response) to 1 (partial response) to 2 (full response), with a score range of 0−66. A higher score indicates a greater ability level18.
The RULM is a validated, SMA-specific outcome measure administered by a qualified clinical evaluator. RULM assesses upper limb functional abilities in young children, non-ambulatory young children and weaker individuals who have a floor effect or a very low score on the HFMSE19. The revised version of the test consists of 19 scorable items: 18 items are scored on a 0 (unable) to 2 (full achievement) scale, and one item is scored from 0 (unable) to 1 (able). The score ranges from 0 to 37 points, with higher scores indicating a greater ability level. The HFMSE and RULM were administered by trained and qualified site clinical evaluators (physical therapist, occupational therapist or national equivalent).
Secondary objectives also included the change in caregiver experience from baseline to week 52, which was assessed using the ACEND instrument completed by the primary parent or caregiver of each participant. ACEND quantifies the caregiver impact experienced by parents/caregivers of children affected with severe neuromuscular diseases, including children with SMA20. The ACEND instrument includes a total of seven subdomains assessing the physical impact domain (for example, feeding/grooming/dressing, sitting/play, transfers and mobility) and general caregiver impact domain (for example, time, emotion and finance); each subdomain comprises several items. Standard domain and total impact scores were generated based on a six-point and five-point ordinal scale associated with the items to conduct a summary analysis. Total scores are meant to convey the amount of ‘impact health’ for each caregiver respondent. Scores range from ‘not applicable’ to 6 for domain 1 and from 1 to 5 for domain 2, with higher scores indicative of reduced impact on caregivers.
An exploratory motor objective included assessment of demonstration of motor milestones according to the Developmental Milestone Checklist containing items from the World Health Organization Multicentre Growth Reference Study21.
Post-hoc analysis
HFMSE scores were analyzed by age at symptom onset, with two groups defined: participants with age at symptom onset ≤6 months and participants with age at symptom onset after 6 months. HFMSE scores were also analyzed for patients at the last visit before receiving add-on therapy.
Data analysis
Results are reported for the FAS, which included all participants who were enrolled in the study and received OAV101 IT. All efficacy and safety analyses were conducted using the FAS. The primary analysis was performed after all participants had completed or discontinued prior to week 52. Categorical data were summarized as frequencies and percentages. For continuous data, mean, s.d., median, 25th and 75th percentiles and minimum and maximum are presented. The analyses were performed using SAS version 9.4 or later (SAS Institute, Inc.).
The number and percentage of participants reporting AEs, related AEs, SAEs and AESI were summarized for the FAS. Summaries were also provided by the Medical Dictionary for Regulatory Activities (MedDRA) system organ class and preferred terms. Change from baseline to week 52 visit in the HFMSE score, RULM score and ACEND instrument total score was summarized descriptively. Exploratory analyses used a mixed model with repeated measurement (MMRM) approach to calculate the LSMs (95% confidence interval) adjusted by baseline age stratum, baseline scores and visits. Motor milestone data were analyzed based solely on the demonstration of milestones at week 52 compared with baseline. No hypothesis testing was performed.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Novartis is committed to sharing with qualified external researchers access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on https://www.clinicalstudydatarequest.com/.
References
-
Wirth, B., Karakaya, M., Jeong Kye, M. & Mendoza-Ferreira, N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu. Rev. Genomics Hum. Genet. 21, 231–261 (2020).
-
Aragon-Gawinska, K., Mouraux, C., Dangouloff, T. & Servais, L. Spinal muscular atrophy treatment in patients identified by newborn careening: a systematic review. Genes 14, 1377 (2023).
-
Kolb, S. J. & Kissel, J. T. Spinal muscular atrophy: a timely review. Arch. Neurol. 68, 979–984 (2011).
-
Genentech. Evrysdi prescribing information. gene.com https://www.gene.com/download/pdf/evrysdi_prescribing.pdf (2025).
-
Biogen. Spinraza prescribing information. spinraza.com https://www.spinraza.com/content/dam/commercial/spinraza/caregiver/en_us/pdf/spinraza-prescribing-information.pdf (2024).
-
Novartis Gene Therapies, Inc. Zolgensma prescribing information. novartis.com https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf (2025).
-
European Medicines Agency. Zolgensma. EMA https://www.ema.europa.eu/en/medicines/human/EPAR/zolgensma (2025).
-
Patel, A. et al. Risdiplam utilization, adherence, and associated health care costs for patients with spinal muscular atrophy: a United States retrospective claims database analysis. Orphanet J. Rare Dis. 19, 494 (2024).
-
Fox, D., My To, T., Seetasith, A., Patel, A. M. & Iannaccone, S. T. Adherence and persistence to nusinersen for spinal muscular atrophy: a US claims-based analysis. Adv. Ther. 40, 903–919 (2023).
-
Day, J. W. et al. Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 22, 632 (2022).
-
Thomsen, G. et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat. Med. 27, 1701–1711 (2021).
-
Meseck, E. K. et al. Intrathecal sc-AAV9-CB-GFP: systemic distribution predominates following single-dose administration in cynomolgus macaques. Toxicol. Pathol. 50, 415–431 (2022).
-
Proud, C.M. et al. Intrathecal onasemnogene abeparvovec (OAV101 IT) for patients with spinal muscular atrophy: phase 3, randomized, sham-controlled, double-blind STEER study. Nat. Med. (2025).
-
Rouault, F. et al. Disease impact on general well-being and therapeutic expectations of European Type II and II spinal muscular atrophy patients. Neuromuscul. Disord. 27, 428–438 (2017).
-
Finkel, R. S. et al. Intrathecal onasemnogene abeparvovec for sitting, nonambulatory patients with spinal muscular atrophy: phase I ascending-dose study (STRONG). J. Neuromuscul. Dis. 10, 389–404 (2023).
-
Krosschell, K. J. et al. A modified Hammersmith functional motor scale for use in multi-center research on spinal muscular atrophy. Neuromuscul. Disord. 16, 417–426 (2006).
-
Krosschell, K. J. et al. Reliability of the Modified Hammersmith Functional Motor Scale in young children with spinal muscular atrophy. Muscle Nerve 44, 246–251 (2011).
-
Glanzman, A. M. et al. Validation of the Expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J. Child Neurol. 26, 1499–1507 (2011).
-
Pera, M. C. et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve 59, 426–430 (2019).
-
Matsumoto, H. et al. Development and initial validation of the assessment of caregiver experience with neuromuscular disease. J. Pediatr. Orthop. 31, 284–292 (2011).
-
Wijnhoven, T. M. et al. Assessment of gross motor development in the WHO Multicentre Growth Reference Study. Food Nutr. Bull. 25, S37–S45 (2004).
Acknowledgements
We thank the investigators, site coordinators and study teams and, most importantly, the patients, families and caregivers for their participation in these studies. We thank clinical consultant J. Salem for his substantive review of the paper. This study was supported by Novartis Pharma, which was involved in the study design, data collection, data analysis, data interpretation and writing of all related reports and publications. Editorial and medical writing support was provided by C. Craige of Kay Square Scientific and D. Wolff of Novartis Pharma. This support was funded by Novartis Pharma.
Author information
Authors and Affiliations
Contributions
Conceived or designed the study: J.V., G.W. and L.Y. Collected data: J.M.K., F.M., L.L.G., K.Y., T.K., C.C., L.D.W., I.R.W., E.M.M., C.M.P., B.T.D., L.H.H., M.O. and W.L.v.d.P. Accessed and verified data: J.V., G.W., A.I. and L.Y. Interpreted data: J.V., G.W., A.I. and L.Y. Wrote or contributed to the writing of the paper: J.M.K., F.M., L.L.G., K.Y., T.K., C.C., L.D.W., I.R.W., E.M.M., C.M.P., B.T.D., L.H.H., M.O., J.V., G.W., A.I., L.Y. and W.L.v.d.P.
Corresponding author
Ethics declarations
Competing interests
Novartis Pharma sponsored this clinical trial. The authors declare the following competing interests. J.M.K. served as a site principal investigator for clinical trials sponsored by Novartis Gene Therapies/Novartis Pharma and Scholar Rock and has received honoraria for serving on an advisory board for Novartis. L.L.G. has received payments for consulting from Roche; served as a member of advisory boards for Novartis; received non-financial support from Roche, Novartis and Biogen; and served as a principal investigator in clinical trials for Novartis, Genethon and PTC. C.C. has received consultancy payments from Roche; has been a non-remunerated member of advisory boards for Novartis, Roche and Pfizer; and has been a principal investigator in clinical trials for Novartis, Sarepta, Biogen, Roche and Scholar Rock. L.D.W. has received speaker and consulting fees from Novartis Gene Therapies/Novartis Pharma, Biogen and Roche; has worked as a principal investigator of SMA studies sponsored by Novartis Gene Therapies/Novartis Pharma, Roche, Scholar Rock and Biohaven; and has received research grants from Novartis Gene Therapies/Novartis Pharma and Roche. She is also a member of the European Reference Network for Rare Neuromuscular Diseases (ERN EURO-NMD). I.R.W. has received honoraria for work performed with Novartis, Biogen, Roche and Pfizer. E.M.M. has received honoraria for scientific advisory boards and educational speaker fees from Biogen, Novartis, F. Hoffmann-La Roche, Sarepta Therapeutics, Santhera Pharmaceuticals, PTC Therapeutics, Pfizer, Scholar Rock and Cytokinetics and has received research support grants from Biogen. His institution receives funding for clinical research from Biogen, Novartis, Roche and Scholar Rock. C.M.P. has served as a consultant for Novartis Gene Therapies/Novartis Pharma, Biogen and Sarepta; served on a speaker’s bureau for Novartis Gene Therapies/Novartis Pharma and Biogen; and received research support from Novartis Gene Therapies/Novartis Pharma, Biogen, Sarepta, PTC, CSL Behring, Scholar Rock and Catabasis. B.T.D. has served as an ad-hoc scientific advisory board member for Novartis Gene Therapies/Novartis Pharma, Merck, Sarepta, Scholar Rock and Roche/Genentech; as a steering committee chair/member for Roche FIREFISH and MANATEE studies; and as a Data and Safety Monitoring Board member for argenX BV and Lexeo Therapeutics. He has no financial interests in any of these companies. B.T.D. has also received research support from the National Institutes of Health/National Institute of Neurological Disorders and Stroke, the Slaney Family Fund for SMA, the Spinal Muscular Atrophy Foundation, Cure SMA and the Working on Walking Fund and grants from Biogen for CS11 and ASCEND studies and from Sarepta Pharmaceuticals, Novartis (Novartis Gene Therapies), PTC Therapeutics, Roche and Scholar Rock. L.H.H. is the site principal investigator for trials sponsored by Novartis, Biohaven, Vertex, Biogen and Astellas. She is also a consultant for Biohaven. F.M. has served as a consultant, scientific advisory board member and site principal investigator for trials sponsored by Novartis, Biogen and Roche. K.Y. and T.K. have no conflicts related to this work to disclose. M.O.’s institution has received research support as a clinical trial site for Novartis Pharmaceuticals, Roche/Genentech and Santhera Pharmaceuticals. W.L.v.d.P. is member of the scientific advisory board of SMA Europe and site principal investigator for trials sponsored by Biogen, Roche, Novartis, Scholar Rock, Biohaven and NMD Pharma. His employer received fees for consultancy service from these companies. J.V., G.W., A.I. and L.Y. are employees of Novartis and own stock/other equities.
Peer review
Peer review information
Nature Medicine thanks Xiaofeng Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor:Jerome Staal, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article
Cite this article
Kwon, J.M., Munell, F., Le Goff, L. et al. Intrathecal onasemnogene abeparvovec for treatment-experienced patients with spinal muscular atrophy: a phase 3b, open-label trial.
Nat Med (2025). https://doi.org/10.1038/s41591-025-04119-2
-
Received:
-
Accepted:
-
Published:
-
Version of record:
-
DOI: https://doi.org/10.1038/s41591-025-04119-2